Leonida Antonio Gizzi, dirigente di ricerca presso l’Istituto Nazionale di Ottica del Consiglio nazionale delle ricerche (CNR-INO), è stato nominato Direttore Scientifico di Extreme Light Infrastructure […]
Leonida Antonio Gizzi, Research Director at the National Institute of Optics of the National Research Council of Italy (CNR-INO), has been appointed Scientific Director of the […]
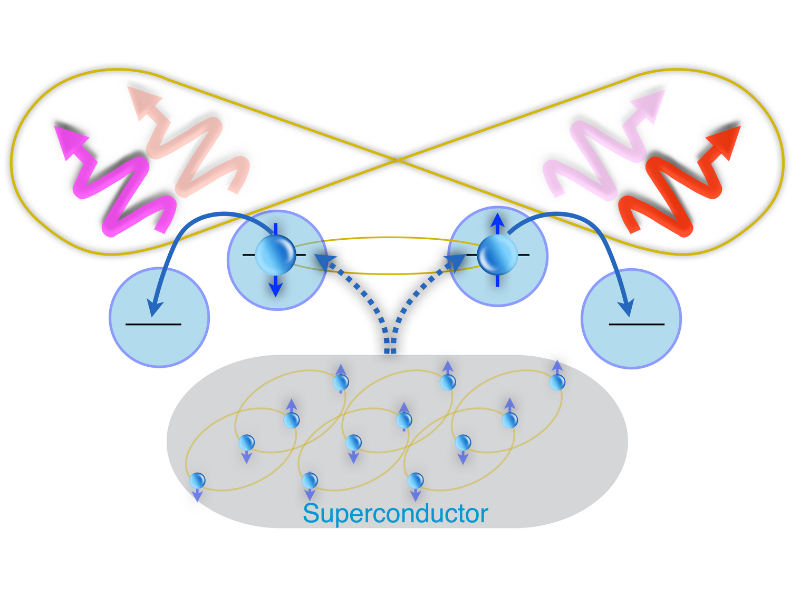
Un recente studio teorico firmato da un team internazionale, che coinvolge anche il CNR-INO, propone un metodo innovativo per generare coppie di fotoni entangled nel dominio […]
A recent theoretical study authored by an international team—including researchers from CNR-INO—introduces an innovative method to generate entangled photon pairs in the microwave regime. Published in […]
Il futuro delle tecnologie quantistiche tra scienza e impresa: Workshop del National Quantum Science and Technology Institute il 20 giugno a Pozzuoli Il National Quantum Science and […]
The Future of Quantum Technologies between Science and Industry: Workshop of the National Quantum Science and Technology Institute on June 20 in Pozzuoli The National Quantum […]
This site uses cookies. If you decide to continue browsing we consider that you accept their use. For more information about cookies and how to delete them please read our Info Policy on cookies use.